●簡國龍/台灣大學流行病學與預防醫學研究所教授
●廖美/中山大學東南亞研究中心助理研究員
●吳介民/中央研究院社會學研究所研究員
(一)時效派觀點
(二)標規派觀點
(三)國產疫苗廠商觀點
(四)爭議重點:疫苗緊急授權、創新臨床試驗、疫情急迫性、產業政策
(五)因應路徑建議
國內 Covid-19 疫情爆發之際,兩支國產疫苗即將完成二期臨床試驗送交審查。5月 28日,疾管署和兩家廠商簽約;政府備受圖利廠商、炒股等質疑。蔡英文總統之前宣布 7 月可以施打國產疫苗;她在 5 月 31 日則公開說明,國產疫苗將以國際科學標準來檢驗。
國產疫苗的研製牽涉科學專業、國民健康、全球人道關懷、國家安全與地緣政治等多個面向,在探討這個議題時,需要通盤思索。本文先從醫學科學的論辯切入,再延伸討論其他議題。

▲疫苗研發必經過試驗環節。(圖/路透)
我們發現,在醫界和公衛界,對於國產疫苗二期如果解盲成功,能否給予緊急使用授權(EUA),當下分成兩派:
一派的觀點堅持,至少要有第三期期中分析(interim analysis)證實保護力才能給 EUA,何況美國 Pfizer、Moderna、Johnson&Johnson,歐洲的AZ也都走這個程序。
另一派的觀點主張,二期試驗結果倘若中和抗體效價夠強,即可視同具有保護力,他們援引發表在《Nature Medicine》的論文為依據,並強調世界衛生組織(WHO)也在研討以「免疫橋接研究」(immuno-bridging study)取代傳統三期臨床試驗的方案。
為了便於討論,我們暫且把前者稱為標規派,後者稱為時效派。我們必須事先聲明,兩派觀點都基於科學證據,都牽涉嚴肅的醫學科學與醫學倫理問題,需要認真看待。以下,讓我們先檢視兩派對問題的討論,最後再提出可行的因應
方案。(相關新聞一、二)
▲疫苗接種。(圖/路透)
(一)時效派觀點
時效派認為在疫情爆發的緊急情況下,應採取權宜措施,不過仍基於科學論證與證據來通過緊急使用授權。他們主張,中和抗體效價可以作為保護力的替代指標(surrogates)。
時效派對國產疫苗深具信心,認為二期解盲成功,就可給予 EUA。時效派也指出 WHO 和 COVAX(見附錄)已經舉辦一系列研討,旨在探討相關議題。
時效派甚至延伸對國產高端疫苗科技的論述,以其蛋白質基底(protein-based)的疫苗株由美國國家衛生院(NIH)提供,而美國 Moderna的 mRNA 疫苗也來自相同疫苗株,隱含其間就有相近的有效性。
然在醫學科技上,蛋白質基底與 mRNA 差異不小。不過,時效派傾向論述,只要二期臨床試驗評估國產疫苗能產生足夠的中和抗體效價,就足以證明其保護力;這樣的說法,乃是根據《Nature Medicine》論文——「Neutralizing antibodylevels are highly predictive of immune protection from symptomatic SARS-CoV-2infection」。
▲疫苗接種。(圖/記者湯興漢攝)
前疾管局局長張鴻仁在談到5月26日WHO的專家視訊會議時指出,在會議中對中和抗體效價和保護力關聯之間曾進行廣泛討論,但會議並未做成結論。
張鴻仁也針對為什麼要替代而不做第三期,引述幾大理由:
一、當全球開始全面接種「第一代疫苗」之後,愈來愈困難進行傳統的以安慰劑為對照的雙盲試驗。
二、全球不能只依賴這幾支疫苗,尤其是中低收入國家冷鏈有問題,mRNA疫苗即便產量夠也不能普及,更何況全球仍供不應求。
三、若能踩在一代的肩膀上快速判定有效的二代疫苗而不為,導致有效的疫苗擺著不用,這明顯不道德。
四、有疫苗可接種的地區,用「安慰劑」做對照組,把受試者曝露在被感染的風險中,有違背醫學倫理的疑慮。
▲疫苗試驗。(示意圖/記者湯興漢攝)
目前擔任疫情指揮中心專家諮詢小組委員的李秉穎認為,「要等第三期,無視於台灣現在緊急的疫情。世界衛生組織為何要開會想要訂保護性抗體標準,就是要讓世界各國疫苗廠有規則可遵循。用血清抗體做效力保證,已經是世界共
識,WHO 都認可。」
他也強調,「新的疫苗若跟目前已上市疫苗比較,如果產生抗體不亞於先前緊急許可授權上市的疫苗,就應該給予緊急授權,但這是緊急授權,不是正式授權。」
前副總統陳建仁近日接受訪問時表示,世界衛生組織召開專家會議探討「免疫橋接」(immunobridging)計畫,乃為了「讓現已完成臨床二期試驗的疫苗公司,只要中和抗體效價和已認證的疫苗一樣好、副作用一樣低,就能取得認證。」

▲前副總統陳建仁。(圖/翻攝自Facebook/陳建仁 Chen Chien-Jen)
然而,WHO 在 5 月 26 日舉行的會議,尚無此結論,與會專家多傾向還是要做第三期試驗,但三期試驗可運用「免疫橋接」概念,設計不同於傳統三期的臨床試驗。關於三期「免疫橋接」的試驗設計,本文在最後回應路徑將提及。
因為國內媒體、各種社媒對這個會議內容的解釋眾說紛紜,有些人選擇特定內容支持自身論點,有些人以訛傳訛,我們建議有興趣者觀看整場會議錄影,便可了解全貌。(Passcode:「JBt*NW49」)
時效派經常引用WHO等機構的會議內容。因此,讓我們看看這些機構最近的重要討論。COVAX「臨床發展和運作特組團隊」(Clinical Development and Operations SWAT team)在6月3日的工作坊會議,進一步整理10家正在執行
三期有安慰劑對照組試驗公司/NGOs 的調查報告。
發現進行三期安慰劑對照組試驗的若干負面趨勢,包括收案比預期落後、有共病性與 65 歲以上群體的收案率特別慢、因血清陽性率增加而導致篩選失敗率較高、還有退出率也增高等問題(註1)。這些新情況,顯示傳統三期臨床試驗的可行性確實需要重新檢視。
▲高端疫苗宣布疫苗二期臨床解盲成功。(示意圖/記者周宸亘攝)
不過,在 5/26 的會議裡,儘管部分與會專家鼓勵納入以「保護力的關聯性」的免疫橋接試驗,作為免除傳統三期臨床的替代方案。但因新冠病毒疫苗產生的中和抗體效價存在幾點基本困境:
(1)檢測並沒有被標準化。
(2)尚未有人類挑戰研究(human challenge study)數據。
(3)也沒有關於抗體和風險的長期數據。(註2)
因為這些困境,也許同一個平台疫苗(比如 BNT 的第一代和第二代)比較問題比較小,但對跨平台疫苗比較(例如在 Moderna 和 AstraZeneca 之間做比較),則短期內因為缺乏數據,難度就較高。此外,現在世界各國陸續出現變種病毒,如何加速核准新疫苗乃刻不容緩的課題。
COVAX 在2021年3月25日舉辦研討會,主要聚焦變種病毒研發改良疫苗等議題,Flores 等人針對變種病毒疫苗的審核途徑指出,免疫橋接(immunebridging)可為沒獲批准原型疫苗的廠商,提供潛在快速生產新的改良疫苗的通道,尤其改良疫苗歸屬在已獲批准原型疫苗的同類(precedented class)中——如Moderna 第一代和第二代。
▲ COVAX。(圖/路透)
他們提出輔助新疫苗快速通道的幾個方針:
(1)接受的可能性取決於新的候選改良疫苗與提供橋接的已獲批准原型疫苗之間的差異度。
(2)需要足夠大的安全性資料庫。
(3)批准後的藥物監控和有效性研究應在計畫引入時就啟動。
(4)在免疫橋接無法被接受的情況下,(a)可用經批准的改良疫苗作為比較基準(comparator),而「非劣性藥效研究」(non inferiority efficacy studies)可能是最佳選擇(按:指改良疫苗的療效是優於或等於現存疫苗作為為控制組的療效);但研究規模可能不可行。(b) 沒有已批准的疫苗對變種有明顯療效,那麼臨床療效試驗設計將取決於流行的病毒株,和現有作為比較基準的已批准原型疫苗的療效;在少數情況下,進行安慰劑對照試驗可能是可行的。(註3)
▲AZ新冠疫苗。(圖/路透)
在目前全球疫情下,第三期臨床試驗的難度愈來愈高(包括倫理問題、人口中血清陽性率大量增加等因素),透過免疫橋接試驗來代替疫苗效力,的確是目前最新疫情下的一個替代方法。
台灣食藥署也使用與上述相同的原則在面對國產疫苗的 EUA。時效派的免疫橋接論點算是亦步亦趨跟著 WHO 的討論,儘管這些討論尚未成為 WHO 官方的正式提案。
註1:這些摘要發佈在 2021 年 6 月 3 日 COVAX 舉辦的研討會:“Booster and Mix & MatchCOVID-19 Vaccine Strategies Planning Ahead in an Environment of Increasing Complexity”所提供簡報,pp.5-7。
註2:Davenport, Miles, 2021. “Immune Protection from SARS-Cov-2.” Presented at WHO meeting on 5/26.
註3:Flores, Jorge, Margaret Toher and David Kaslow, 2021. “Pathways for Approval of COVID19 Vaccines Based on SARS CoV 2 Variant Strains,” presented on “SARS-CoV2 variants
Practical considerations for accelerated clinical development in light of current regulatory
guidance,” Clinical Development & Operations SWAT Team, sponsored by CEPI, GAVI, WHO,
and UNICEF, March 25.
▲聯亞指出,一般三期臨床試驗是在疫情嚴重的地區做,要在台灣施行的難度很高。(圖/記者蔡玟君攝)
(二)標規派觀點
標規派主張者中,台大醫院臨床試驗中心主任陳建煒很具代表性。他在今年三月寫的一篇文章廣為流傳:
「施打疫苗後,人體的免疫系統會產生中和抗體及許多的細胞反應來對抗病毒,在這些對抗病毒的武器中,中和抗體比細胞反應容易測量,故被視為一個替代的療效指標(surrogate end point),然而『施打疫苗、有產生中和抗體』就等於『捉到老鼠』了嗎?
應該不是!更重要的是能預防有臨床症狀的新冠肺炎,特別是重症(要住加護病房、用呼吸器、葉克膜或死亡);因此先進國家如英國、美國及歐盟的藥政管理單位核准疫苗緊急使用授權(EUA)的條件,並不是中和抗體的產生,而是必須看到疫苗能有效地預防有臨床症狀的新冠肺炎。
…… 醫學文獻及醫療產品發展過程中『理論上有效、初期人體試驗有效,但大規模臨床試驗發現是無效或有害』的案例太多了,再具有吸引力的理論,還是需要大規模臨床試驗加以驗證,這才是近代實證醫學的精神。」
最近,陳建煒重申應以三期試驗期中分析為基準:
「從 2020 年底至今,先進國家及 WHO 核准緊急使用的新冠肺炎疫苗,都是用三期試驗的期中分析數據、實證支持可預防臨床疾病為基準,台灣要用更寬鬆的標準,來給國產疫苗緊急授權不是不可以,但應該清楚說明,我們核准的標準是什麼?不同專家對於這課題有不同意見,公開透明才能取得科學社群的信任。」、「新冠肺炎是新興疾病,和流感、A 型肝炎等已研究多年的疾病不同,雖然很慶幸有數個新冠肺炎疫苗已研發成功,但中和抗體數據仍在分析中,現在不宜妄下結論。」
▲新冠肺炎疫苗研發。(示意圖/路透)
基本上,「相同病毒株」是援引生物相似性(biosimilarity)的原理。免疫橋接試驗是看產生抗體的生物相似性反應,但除了抗體以外,臨床保護力仍需考量T細胞(T-cell)的反應。
時效派強調免疫橋接,但對不同廠牌(平台)的疫苗,國際標準仍需有臨床療效(clinical outcome)指標。美國FDA在去年十一月底核准Pfizer/BNT疫苗的EUA公告,第六頁執行摘要(executive summary)有詳述參與三期臨床試驗的人數和效力及安全性判定的標準,均超過三萬餘人。
美國 FDA的緊急授權的確依據期中分析的結果,這大概是「未完成三期臨床試驗」的說法來源,但若講成「未經臨床三期試驗就批准 EUA」,則是誤導。第三期臨床試驗進行期中分析是為考量療效及安全性的監測會議,明訂提早結束之規定(stopping rule),由廠商提給 FDA。
假若不走正規三期路徑,除了免疫橋接試驗,還有「Non inferiority trials withdisease endpoint」(非劣性試驗)可操作,就是在三期試驗中,對照組不用placebo,而以既有 EUA 的疫苗作為對照組,看新疫苗的保護力是否不亞於既有
疫苗(new vaccine is not appreciably worse than licensed vaccine),如果過關,即可EUA。這個提議由Dean Follmann 在 2021 年 1 月 28 日 COVAX 的研討會中提出。(註4)
▲台灣疫苗短缺。(圖/路透)
上述 COVAX 在今年 3 月的研討會議也如此建議。若採取非劣性試驗研究路徑,就可以避開時效派所說「在盛行區施打安慰劑不符合倫理的難題」。美國政府在 SARS、MERS、Ebola、茲卡病毒爆發後,就檢討過個人防護設備(PPE)不足以作為控制的方法,而朝向發展疫苗。
2017 年聯邦制定新法規,基於國安理由,NIH 鼓勵開發生物製劑,包括 mRNA 研究。Covid-19爆發之後,FDA曾經對EUA指導原則做過數次調整,分別是2020年10月、2021年2月22日、2021年5月25日(也是最近一次)。2020年10月調整時,有針對核准EUA的標準做若干修改。
但是,後來美國幾支新冠肺炎疫苗獲得 EUA,都採取正規程序:亦即,都是三期期中報告通過 FDA 審查,而獲得 EUA,其中 Pfizer、Moderna、 Johnson & Johnson 都走完這個程序。
雖然,採用中和抗體效價作為保護力相關性的指標,在其他疫苗都有實例,例如 B 肝疫苗和流感疫苗,這乃使用生物相似性的原理。但是新冠肺炎是新的疾病,恐難直接套用,因為單是 T-cell 反應的定量為何,就沒有足夠的資料,更遑論其他。
註4:4 P. 32 in “Licensure of New Vaccines Going Forward,” presented by Dean Follmann on “Emerging Challenges to the Development of Covid 19 Vaccines,” Clinical Development & Operations SWAT Team, January 28,2021sponsored by Covax, CEPI, GAVI, and WHO.
▲新冠病毒。(示意圖/取自免費圖庫Pixabay)
(三)國產疫苗廠商觀點
現在,讓我們看看國產疫苗廠商的觀點。目前,國內兩家疫苗廠都在二期收尾階段,預計於六月中下旬提交二期臨床試驗報告。(註5)
本土藥廠主張,做完「擴大二期」就會申請 EUA,是根據衛福部食藥署在 2020 年 10 月訂定的規定:「食藥署在去年 10 月底公布台灣 EUA 標準,將二期人數從數百人擴增至 3500 人(莫德納二期臨床人數是 600 人),再加上 1 個月安全觀察期,以超過二期人數的『類三期』方式通過。為何是 3500人設計?據食藥署說法,是參考 WHO、美國 FDA 指引,『符合國際標準精神訂定出來的』。」

▲聯亞生技營運長彭文君。(圖/記者林育綾攝)
聯亞生技營運長彭文君表示:
「聯亞的疫苗第二期臨床試驗做到了 3850 人,但本來規劃是打算比照國外,臨床一加二期做幾百人趕快結束,進入臨床三期大規模試驗。」
「去年醫藥品查驗中心(CDE)、食藥署開很多專家會議,決議要臨床一期做完後做大規模臨床二期試驗,『我們是配合政府』,聯亞生技原本臨床規劃非如此,是專家會議達成共識後,政府要求比照做才能取得EUA。」、「台灣的感染人數跟國外比太懸殊,不具客觀條件做臨床三期,所以是做『準三期』,這與 WHO 方向一致。」
事實上,當全球疫情爆發初期,國家衛生研究院曾在去年 4 月召開關於「疫苗緊急開發策略」會議,接著在 7 月和 11 月又召開其他兩次會議,分別探討疫苗研發及緊急使用授權和疫情挑戰及疫苗產業等議題,這三次會議都有廠商參
與。
最後一次專家會議提出建議包括「國內廠商通過台灣 EUA 審查後,若國外已經公布新冠疫苗的臨床保護力的關聯(correlate of protection, CoP),國內可根據 CoP 規範來進行疫苗使用核准授權疫苗正式上市;若國外尚未公布 CoP,政府應輔導國內廠商進行跨國第三期臨床試驗,證明疫苗的臨床保護力,協助國內疫苗廠拓展國際市場。」
▲國產疫苗如何擠身國際?(圖/路透)
所以,這個當時集結部分標規與時效兩派專家的會議,對國外如果沒有公布 CoP 標準,政府基本上應該輔導廠商進行三期。但是,我們發現,從上述疫苗廠商的認知看來,廠商主張二期解盲成功即可向食藥署申請 EUA 乃根據法規。廠商的這個認知,與上述國衛院專家會議的結論是否符合,仍須商榷。
而目前台灣疫情居高不下,外購疫苗在短期內尚不足以支應國內需求,在此情況下,蔡總統表示國產疫苗將可在七月施打,這是一個政治宣示。而食藥署也必須依法規而行,根據已公布 EUA 標準進行審查。
現在時效派醫界人士主張二期解盲成功即可考慮 EUA,而標規派堅持美國醫學科學的標準,兩派爭持不下。因此,在可見未來,關於國產疫苗的緊急授權,很可能造成一場輿論風暴。政府不能不對此定時炸彈預先防範。
註5:高端疫苗的二期試驗,林奏延是計畫總主持人;二期試驗分成 7 組,低中高劑量各2 組、placebo 1 組。

▲我國新冠肺炎疫苗EUA審查標準。(圖/翻攝自衛福部臉書粉專)
(四)爭議重點
(1)關於疫苗緊急使用授權
標規派認為必須按部就班,按照歐美既有程序才能緊急授權,如果「二期解盲成功就緊急授權」有很大風險。時效派則認為二期解盲成功就可緊急授權,以中和抗體效價來代替測量保護力具有科學根據,也在 WHO 相關研討會多次討論;而且在目前疫情情況下,三期試驗不但執行困難也緩不濟急。
因此,免疫橋接可以取代傳統三期臨床試驗。但標規派認為,關於免疫橋接在 WHO 尚無共識。我們認為,如果 WHO 和 COVAX 等組織能夠整合足夠的免疫橋接相關研究,累積更多臨床試驗資料,而擬定擬定新規範,便能讓標規派與時效派的觀點獲得調和,畢竟本質上,這兩派還是基於科學證據在堅持其主張。
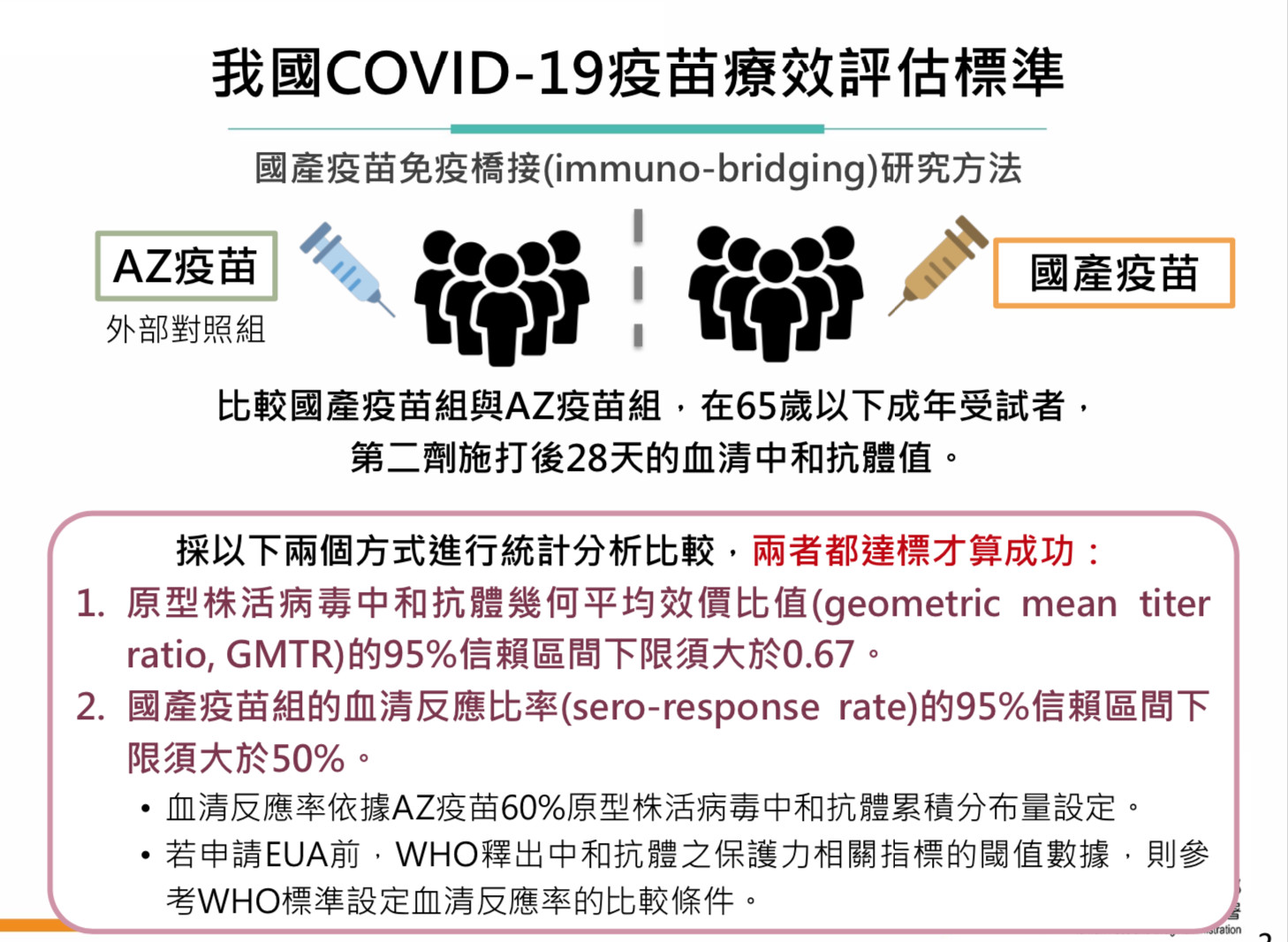
▲國產疫苗8月開打?療效與AZ比,未通過「2必要條件」不核發EUA。(圖/食藥署提供)
(2)創新臨床試驗
全球疫情尚在蔓延之中,尤其新的變種病毒正在許多發展中國家肆虐。雖然在大部分富裕國家因為大量施打疫苗而控制疫情,但在許多無法大量獲取疫苗的國家情勢仍然極為緊急。
在此背景下,WHO、Covax 等組織密集研討以創新的試驗程序取代傳統三期試驗,就顯示其緊急救援的人道意義。這些被提出的創新三期試驗,包括了「免疫橋接試驗」、「非劣性臨床試驗」、「免疫原性比較試驗」等等。
這些創新試驗,具有未知的風險;創新的目的是在提供更多快速、安全而相對低廉的疫苗。我們對這些創新試驗,應該保持開放而審慎的態度,因為在緊急疫情下,人類必須兼顧標規與時效才能妥當處理我們所面臨的巨大危機。
▲WHO、Covax 等組織密集研討以創新的試驗程序取代傳統三期試驗。(示意圖/路透社)
(3)全球疫苗供應急迫性
疫苗是戰略物資,除了國民健康,也牽涉國家安全,尤其台灣外交處境艱難,更需要掌握國產疫苗進度。目前第三世界嚴重缺乏疫苗(因此讓中國和俄國的疫苗外交奏效)。
時效派認為,如果能讓第二代的疫苗以新規範儘快上市,將有助於緩解台灣疫情。國產疫苗是否儘早緊急授權,必須與外購疫苗與外國贈與進度做通盤考慮,日本已經給予台灣 124 萬劑AZ,美國承諾 75 萬劑,後續是否繼續提供應列入衡量。
若疫苗外購與贈與進度順利,可為國產疫苗進度爭取較多時間,讓我國監管機構有更多餘裕進行審查,不要讓急切心態模糊了整體疫苗供應評估。若將來台灣能以國產疫苗協助世界其他地區,也有助於緩和疫苗供應的全球南北差異,讓「Taiwan can help」與時俱進。
(4)產業政策問題
在國家安全和發展政策(扶植策略產業)下,世界強權國家加緊研發,提早緊急使用授權,有如醫療科技競賽。美國、歐洲、中國與俄羅斯莫不如此。
俄中兩國一些疫苗都是在二期後就針對特定群體開打(因此都曾被質疑試驗過程不透明),但獲得 WHO 批准的中國國藥疫苗(Sinopharm)有經過三期;WHO 最近又核准中國科興疫苗(Sinovac)。
▲中國科興疫苗。(圖/CFP)
過去台灣產業發展都是按部就班,跟隨先進國家(尤其是美國)規範走。台灣目前生技基礎研究能力尚在努力追趕美國 FDA 要求的標準。
如果這次要繞開美國既定規範(如標規派所言),在審查程序上要特別慎重。由於目前國產疫苗陷入炒股質疑,政府在執行產業政策和監管上也須特別小心,絕對要避免落入「圖利廠商」、「尋租」等泥淖而傷害政府公信力與產業政策正當性。
從更大格局討論,國產疫苗最終目標是要進入國際市場。除了美國規範,我們也應該關注其他國家與國際機構的規範及其調整趨向,因為國產疫苗若研發成功,將來推向國際市場需要各國政府和國際組織認證。
因此,尋求 WHO、EMA(European Medicines Agency)、ACCESS Consortium(註6)等機構認可,仍是重要步驟;政府應該在這方面積極輔導廠商或給予協助。
註6:Access Consortium 由澳洲、加拿大、新加坡、瑞士、英國等五國監管機構組成。
▲AZ疫苗。(圖/路透)
(五)因應路徑建議
以下第一點與第二點是不同的政策選擇的路徑。我們保持開放的態度,但提醒政府在做不同決定時都具有風險與成本(法律、社會、政治等面向),必須謹慎斟酌。第三到六點則是延伸的路徑建議。
(1) 如果政府決定國產疫苗須走目前美國標準的三期臨床試驗程序,則廠商可在三期執行過程中提出期中報告,報告審查通過即可給予 EUA。但這個程序將會使國產疫苗施打計畫可能至少再延長半年,如何解決疫苗短缺問題,也必須提出配套措施。
(2) 政府若決定以二期報告作為國產疫苗緊急使用授權的依據,必須了解這是「在民主國家中的監管規範創新」,因為歐美民主國家目前尚未在在二期結束就給予緊急授權的先例,而且 WHO 組織及其專家會議也尚未達成共識。
因此,政府需要以開放透明態度向國人告知此乃緊急情況下的決策依據。同時,如何在醫界兩派爭執之中求取共識也是當務之急。
政府必須清楚告知國人,採取二期後緊急授權的「新規範」有一定風險要承擔,包括針對疫苗副作用和安全性的追蹤;藥廠與監管機構不斷累積有關療效的數據。在這條路徑中,政府必須步步為營,預先規劃處理風險的步驟與方法。

▲接種疫苗多少都會有副作用。(示意圖/記者黃克翔攝)
(3) 國產疫苗二期審查後是否給予緊急授權,都可持續進行三期試驗。如果通過二期審查,可採取多國多中心的方式,執行第三期的非劣性臨床試驗,以國產疫苗作為實驗組,以既有 EUA 的疫苗(AZ、Moderna 等)作為對照組。
這樣不但可以處理疫苗短缺,也可以解決「在盛行區施打安慰劑不符合倫理的難題」。再者,政府也應促成國產疫苗廠商與國際大藥廠合作,如此進行的多國多中心臨床試驗,更能獲取信度高的樣本。
(4) 國產疫苗可以考慮最近 WHO 會議提出的一個有啟發性的三期臨床試驗:在第三期次群體樣本進行免疫橋接研究,包括在部分樣本中以及對罹病者收集血清作兩階段測量:分別在第 1 天及第 29 天為第一階段,作初級抗體反應分析;而自 71 天到 728 天為第二階段的持續抗體分析,藉以檢驗抗體效價和臨床療效的相關。
(5) 無論選擇上述(1)或(2)方案,國產疫苗廠商可以和國際大型藥廠及知名受託研究機構(Contract Research Organization, CRO)合作執行三期試驗。這個方案可以和(3)和(4)方案同步考慮。(註7)
▲國產疫苗廠商可以和國際大型藥廠及知名受託研究機構,合作執行三期試驗。(示意圖/路透)
(6) 政府鼓勵國產疫苗的目的,除了國民健康、國家安全的考量之外,也具有鼓勵本土產業發展的用意。疫苗產業要能夠切入全球疫苗供應鏈,涉及化學製造管制(Chemistry, Manufacturing & Controls, CMC)等流程,因此協助疫苗廠商走向國際市場,將是未來政策重點。
政府可以協助國產疫苗獲得國際機構認證,將國產疫苗產業推向國際,讓施打國產疫苗獲得通行國際的疫苗護照。
註7:
假如台灣疫情狀況不適合做三期,而三期研究非常昂貴,動輒數億美元,而且我國的國際政治情境可能也比較難在國外做大規模疫苗試驗,那麼台灣可以採取的策略是與國外大藥廠合作,假如二期解盲後,國產疫苗真的中和抗體效價很好,品質優良,可與國外藥廠採取合作、策略結盟等方式,在其他盛行率高的國家進行三期。
如果保護力強,半年內就可提出期中報告,申請歐美國家和 WHO 的緊急授權,這樣同時也可以打開國產疫苗的國際市場,既符合扶持國安策略產業(符合國家利益),又具可行商業模式(符合開發疫苗藥廠的利益)。
與美國大藥廠合作的好處是可以利用其資金優勢,找大型 CRO 做三期試驗。一般國際大藥廠(sponsor),三期都是委託給 CRO(contract research organization)公司,CRO 依循臨床試驗標準流程,做完三期,向 FDA申請 NDA (New Drug Application) 的流程。
▲政府可以協助國產疫苗獲得國際機構認證,將國產疫苗產業推向國際。圖為莫德納疫苗(圖/記者周宸亘攝)
附錄
COVAX(COVID-19 Vaccines Global Access 嚴重特殊傳染性肺炎疫苗實施計劃)在 2020 年上半年成立「臨床發展和運作特組團隊」(Clinical Development and Operations SWAT team),專門處理臨床開發和運作等相關議題,包括支持臨床試驗場域的準備和建立其間網絡,也研討疫苗安全和疫苗科學等。
目前關於新冠肺炎最前沿研討,多半由這個團隊先透過工作坊發動議題,再醞釀成為世界衛生組織和其他國家的共識。這個工作坊每月定期開一次會,關於開會主題、會議報告、工作坊討論素材的 PPT、工作坊的問與答,或是會後論壇,都發佈在其專屬網頁。建議關注相關議題的政府單位、廠商、研究者、媒體、與大眾上網查詢。
當我們在閱讀網站下載文件,會發現封面有流行病防範創新聯盟(CEPI)、疫苗聯盟(Gavi)、世界衛生組織(WHO)、聯合國兒童基金會(UNICEF) 的標誌,乃緣於COVAX 最早由上述組織發動組成,目前有 192 個國家參與其中;而「臨床發展和運作特組團隊」隸屬 COVAX ,不過每次參與工作坊討論的專家則根據議題時有變動。

▲COVAX 最早由流行病防範創新聯盟(CEPI)、疫苗聯盟(Gavi)、世界衛生組織(WHO)、聯合國兒童基金會(UNICEF) 組織發動組成。(圖/路透)
熱門點閱》
► 國產疫苗解套良方:生物相等性第三期臨床試驗(陳秀熙、許辰陽、張維容、林庭瑀、嚴明芳、陳立昇、任小萱)
●本文獲作者授權,轉載自「吳介民」臉書。以上言論不代表本網立場,歡迎投書《雲論》讓優質好文被更多人看見,請寄editor88@ettoday.net或點此投稿,本網保有文字刪修權。





我們想讓你知道…國產疫苗的研製牽涉科學專業、國民健康、全球人道關懷、國家安全與地緣政治等多個面向,在探討這個議題時,需要通盤思索。本文先從醫學科學的論辯切入,再延伸討論其他議題。