●許辰陽/台灣大學公共衛生學院公共衛生碩士學位學程兼任助理教授
●張維容/台灣大學公共衛生學院預防醫學與流行病學研究所博士
●林庭瑀/台灣大學公共衛生學院預防醫學與流行病學研究所博士
●嚴明芳/台北醫學大學口腔醫學院口腔衛生學系教授
●陳立昇/台北醫學大學口腔醫學院副院長
●任小萱/台灣大學公共衛生學院預防醫學與流行病學研究所博士後研究員
●陳秀熙/台灣大學公共衛生學院預防醫學與流行病學研究所教授
2020新冠肺炎疫苗臨床試驗科學
任何疫苗或藥物的發展與上市使用,都需經過完整的臨床試驗科學評估過程,因為疫苗是要接種於人體上,其安全性與有效性成為相當重要的議題。疫苗的研發必須先利用動物,檢視疫苗接種後,其免疫與生物反應的臨床前期先驅研究之後,才能進行一至三期臨床人體試驗及第四期上市監測。
第一期的人體試驗,主要是測試少量試驗者(大約10-20位)對疫苗的劑量、免疫原性(immunogenicity)與安全性。第二期的人體試驗則會擴大(規模至少百人)測試疫苗的安全性及免疫原性,以此擴大參與族群,並以科學上客觀的隨機分派試驗盲測設計方法(一組為實驗組,另一組為對照組),檢視疫苗的安全性和刺激免疫系統,產生保護感染中和抗體濃度在兩組間之比較。
之後進到第三期的人體臨床試驗,擴大族群至數萬人,除了再次檢視接種的安全性外,並使用臨床隨機分派試驗設計,檢視接種者與未接種者的長期效益包括症狀個案、重症及住院減少,以評估疫苗之保護力。
▲國產疫苗遭質疑未經「三期臨床試驗」。(圖/記者蔡玟君攝)
因此,世界各國及WHO在核準疫苗上市前都會審查上述的完整試驗結果,再由各國FDA決定是否批准該疫苗之使用。此過程通常需要耗時多年才能上市。而新冠肺炎很快在2020年內經研發、量產及上市來成為對抗新冠肺炎的最佳利器,而過程中當然就必須經過歐美國家緊急授權使用(Emergency Use Authorization,EUA)。
新冠肺炎疫苗在2020年3月中WHO宣布世界大流行之後不同種類疫苗開始進行實證醫學臨床試驗第一、二及三期,其中包括mRNA、載體及重組蛋白。
目前全世界超過122個新冠病毒(SARS-CoV-2)疫苗在研發中,有83個疫苗進入第一、二期人體試驗中,34個疫苗正進行第三期人體試驗,17種疫苗己完成人體試驗並受到至少一個國家授權,這些疫苗包含大家熟知如美國的輝瑞、莫德納、英國的牛津疫苗、以及嬌生疫苗,在科學實證的支持下完成疫苗開發並進行量產。
目前發表於具科學公信力國際學術期刊的第三期臨床試驗顯示,輝瑞疫苗與莫德納疫苗對於新冠肺炎的保護力可達95% (95%信賴區間: 90-98%,Polack et al., 2020)與94% (95%信賴區間: 89-97%,Baden et al., 2021),英國的牛津疫苗與嬌生疫苗的保護力則分別可達71% (95%信賴區間: 50-94%,Voysey et al., 2021)與66.9%(95%信賴區間: 59.0-73.4%,Sadoff et al., 2021)。
▲輝瑞疫苗。(圖/路透)
疫苗臨床試驗緊急授權(EUA)科學與倫理兩難
由於疫情非常嚴峻,為了能夠及早施打疫苗降低疫情,常針對第三期臨床試驗期中報告(Interim Report)以準予量產成為可以正式上市施打疫苗。然而這樣提前授權在科學與醫療倫理掀起兩難抉擇。
(1)倫理面
從倫理角度來看,既然期中以經有了良好結果之跡象,安慰劑組是否可以提前施打疫苗來降低得到新冠肺炎個案的風險。
(2)科學面
若是如上述安慰劑組在第三期臨床試驗施打疫苗則可能會產生因為對照組汙染(control contamination)而對於長期效益結果(如症狀個案、重症、住院及死亡降低)評估產生影響。
由於上述科學以及倫理兩難情況下,對於未來各國新開發疫苗,是否可以援用歐美國家上述利用EUA方式引起各界不同爭議。如何使用更有效的方法來解決兩難之議題,是任何新冠肺炎疫苗第三期臨床試驗所面臨挑戰。
▲新冠肺炎疫苗第三期臨床試驗需面臨多項挑戰。(圖/路透)
疫苗與變種病毒競爭
另一個是科學與變種病毒之競爭難題,早期疫苗的開發多針對武漢演化株或D614G病毒株為主要目標開發疫苗,在此一情境下開發的疫苗所激發的人體免疫原性也是針對早期的病毒株,這些保護性抗體在目前變種病毒在世界各地頻傳並且逐漸成為主流病毒的情境下,也面臨需要改良既有的疫苗之需求。
因此,為達到有效率的開發與改良新冠肺炎疫苗,解決世界上對於疫苗的廣泛需求,以及改良舊有疫苗對於變種病毒具有保護力,目前亦有運用二期疫苗施打的免疫反應,所產生中和抗體濃度在新舊病毒比較相等性橋接第三期臨床試驗,成為另一個以替代評估終點佐證的科學方法。
但此種作法仍然會有各國新開發疫苗,未曾進行舊有病毒株第三期臨床試驗之議題,雖然目前已經不可能再進行舊有病毒株第三期臨床試驗,對於國產疫苗開發之科學評估,仍必須找出方法突破困境進行即便在橋接過程中可以滾動式方式進行第三期臨床試驗。
從醫療倫理面觀來看,在目前疫苗可以取得的情形下,仍以安慰劑建立第三期臨床試驗的對照組也有可議之處(Cyranoski, 2020)。
▲新冠肺炎變種病毒。(示意圖/路透)
疫苗效益生物相等性研究設計
為能夠解決國產疫苗第三期臨床試驗科學困境,以台灣而言,目前已有國外疫苗(主要以AZ牛津疫苗為主)在疫情流行間進行施打,可以藉此使用國產疫苗和AZ疫苗進行生物相等性(Non-inferiority)第三期臨床試驗。其基本原理為:
(1)AZ疫苗效益:AZ疫苗組>安慰劑組(第三期臨床試驗已證明及核準)
(2)國產疫苗效益:若國產疫苗組≈AZ疫苗組
(3)推論:國產疫苗組>安慰劑組
如此可以避免使用安慰劑組之宭境。因此,如何以有效率的研究設計運用第三期臨床試驗,以建立國產疫苗科學實證基礎,在目前台灣疫情情境下,應該可以此種生物相等性第三期臨床試驗彌補在橋接二期至三期變通辦法中,同時達成第三期臨床試驗的標準。
這樣第三期臨床試驗可如圖一設計,為提高效率可選擇接觸確診個案之可疑感染者(如接觸的家戶成員、任何群聚感染後及高危險群接觸者),進行雙盲隨機分派再比較兩組長期效益。
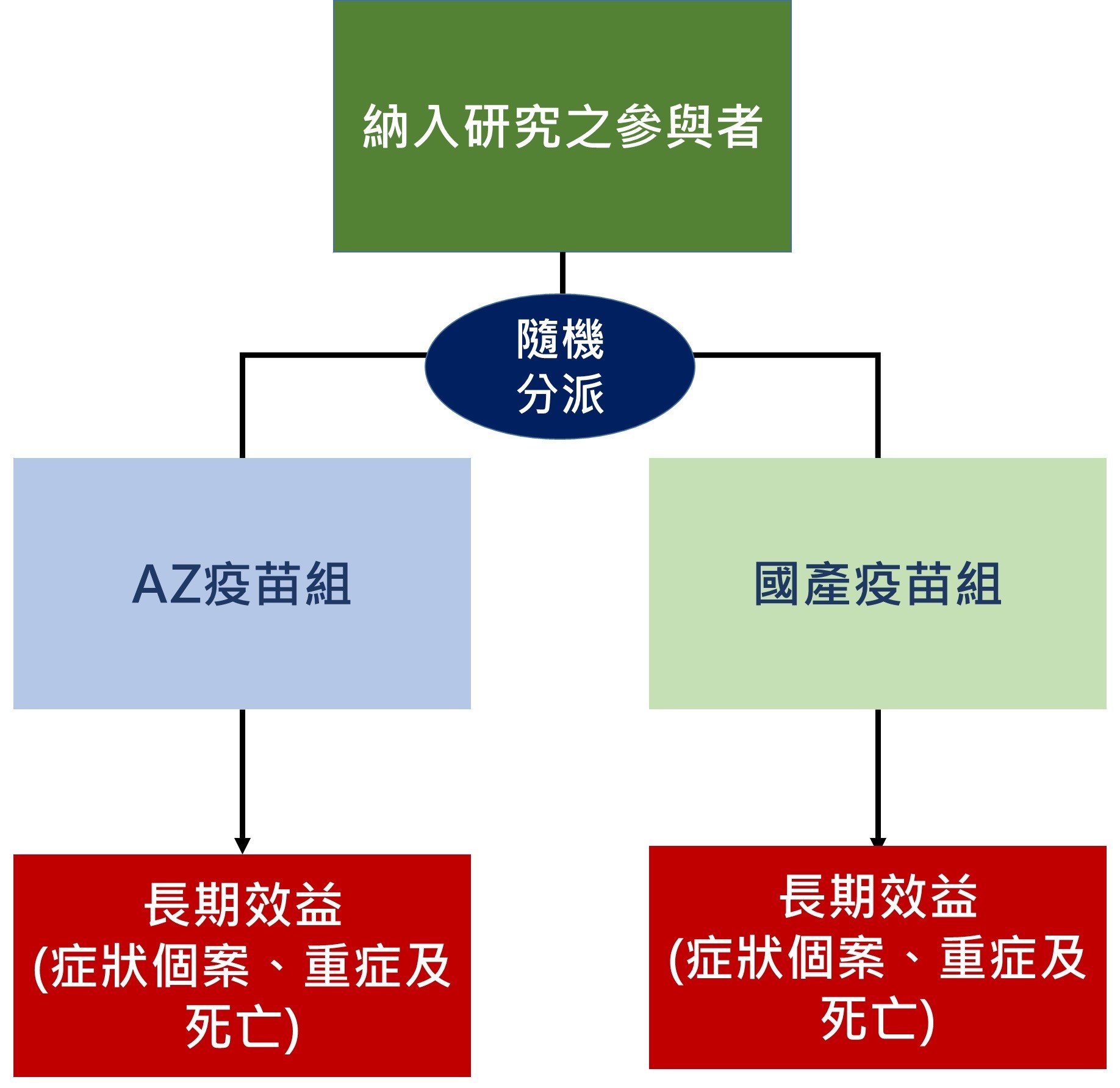
▲圖一、新冠肺炎國產疫苗生物相等性第三期臨床試驗研究設計。(圖/撰稿團隊提供)
疫苗效益生物相等性的樣本數估計
以此種第三期疫苗生物相等性試驗性質來說明,若國產疫苗在試驗結果的罹病風險低於或等同於AZ疫苗,就證明國產疫苗也可以如AZ疫苗可廣為人群使用。由於此研究設計不同於傳統以安慰劑組作為比較基礎,因此研究進行中最為重要的樣本數估算也需要精準。以下運用先前科學研究所提出的樣本數估計方法,評估此一生物相等性研究設計所需納入的參與人數(Farrington and Manning, 1990)之可行性。
運用前述之隨機分派研究設計可以估算兩疫苗組罹病率差距,若兩疫苗的保護力相當,則可以預期AZ疫苗組的罹病率與國產疫苗組的罹病率將非常相近,可以寫為:
兩組罹病率差距= AZ疫苗組罹病率 – 國產疫苗組罹病率
若兩組罹病率差距小於預先設定的數值,則我們即可以推斷國產疫苗的保護效果至少與AZ疫苗相當。在此一效益評估過程中,兩疫苗組保護力的差距需與利用實證資料取得估計的不確定性比較,以降低推斷過程可能產生偏誤的可能。
若兩組納入的研究人數越多,則罹病率差異估算的過程之不確定性就越小。因此運用Farrington and Manning方法,結合文獻報告的AZ疫苗罹病率(每千人5.2,Voysey et al., 2021)與預先設定的可接受的5%型I誤差,以及統計檢定力(80%)即可以在研究執行前估算兩組所需納入的人數。詳細樣本數估計公式見附錄。
▲AZ疫苗。(示意圖/記者湯興漢攝)
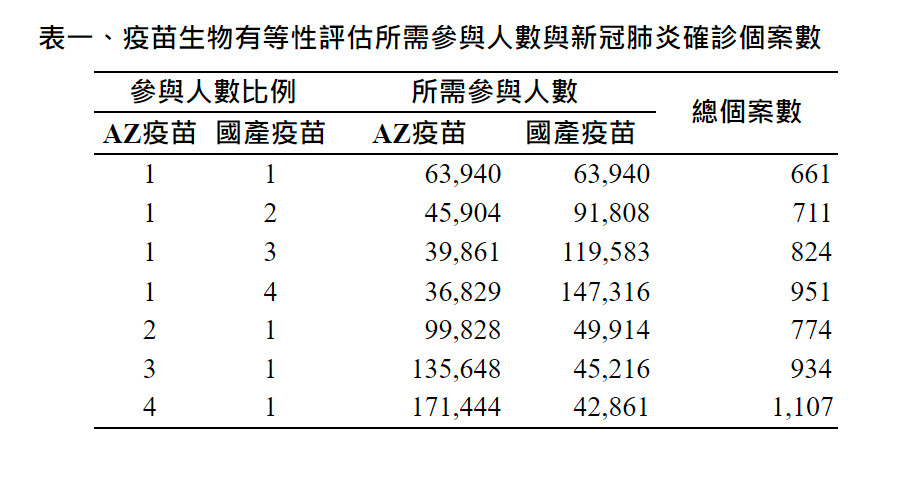
表一為在兩組納入參與者人數比例由1:1依序到1:4與4:1達到生物有等性評估所需的總人數,以及新冠肺炎確診個案數。由表中可見,在1:1之參與比例下,所需之確診個案數共661人,約為目前全台灣2天之確診人數。
所需之參與人數共約13萬人,遠少於目前可供給之疫苗劑數。即使在4:1之參與比例下,所需之確診個案數為1107人,約為目前台灣4天之確診人數,總參與人數則為21萬人,亦遠低於目前疫苗可供應劑數。此顯示在台灣目前疫情仍然有持續群聚感染情況下,且疫苗大量獲得之前,可以趕快進行此相等性且不需要安慰劑組的第三期臨床試驗。
以生物相等性研究設計橋接第二期與第三期臨床試驗
延續此生物相等性研究設計以及樣本數估算,此研究設計也可涵括日後橋接第二期與第三期連床試驗所需之估算。在第二期試驗中已經對於參與者皆進行完整之檢測評估,包含抗體濃度監測。
完成此施打後之濃度即可提供做為二期橋接第三期臨床試驗之評估,不需追蹤等待參與者之罹病、住院以及死亡之資訊收集。此情況特別適用於當此第三期生物相等性試驗進行時,若遇到疫情趨緩及受控制時之情形。
重要的是此第二期抗體濃度間比值也可以提供第三期生物相等性臨床試驗之樣本數的精算。以AZ疫苗發佈之中和抗體濃度與假設可以在解盲後得到國產疫苗抗體濃度相比推算(Folegatti et al., 2020),在橋接評估中兩組所需之參與者比例為1(AZ):1.29(國產疫苗),所需之參與者為128,097人(AZ:55,986人,國產疫苗:72,111人),總共需662位新冠肺炎確診個案。
結論
目前全世界以及台灣皆面臨發展疫苗應付新型變種病毒,在緊急授權造成的科學及倫理兩難情境下,運用所提出之第三期生物相等性臨床試驗,不僅可作為二期橋接三期變通做法在科學上的解套,且不需要安慰劑組符合台灣民意風情,也可使國產疫苗成為紮實實證醫學產品受到國際間認定,達到長程發展台灣自己疫苗生技產業。
附錄:樣本數估計
今欲進行一生物相等性臨床試驗證實兩疫苗同樣有效,將受試者隨機分派在暨有疫苗組(以牛津疫苗為例)及國產疫苗組,則兩組的罹病個數皆服從二項分布且各自有其疫苗施打後罹病率。統計假說設立如下:
虛無假說:國產疫苗罹病率-牛津疫苗罹病率-臨界值≥0
對立假說:國產疫苗罹病率-牛津疫苗罹病率-臨界值<0
若兩組疫苗具有生物相等性,則其疫苗效益差異為0,此時我們會設立一個可以接受的臨界值,如20%。
另外,我們進一步設立兩組樣本數比例,若兩組樣本比例為1:2,亦即在一個假設總計要招募135,000個參加者的試驗,則需要分派45,000及90,000名至牛津及國產疫苗,在此利用兩個疫苗在第二期臨床試驗中和抗體濃度比進行橋接樣本比例,則總計所需要的樣本數(N)估計公式如下:
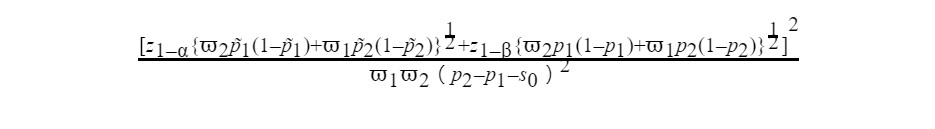
本文中其他參數值如下表所示:

參考資料
Baden, Lindsey R., et al. "Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine." New England Journal of Medicine 384.5 (2021): 403-416.
Cyranoski, David. "WHY EMERGENCY COVID VACCINE APPROVALS COULD POSE A DILEMMA." Nature (2020): 18-19.
Farrington, Conor P., and Godfrey Manning. "Test statistics and sample size formulae for comparative binomial trials with null hypothesis of non‐zero risk difference or non‐unity relative risk." Statistics in medicine 9.12 (1990): 1447-1454.
Folegatti, Pedro M., et al. "Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: a preliminary report of a phase 1/2, single-blind, randomised controlled trial." The Lancet 396.10249 (2020): 467-478.
Polack, Fernando P., et al. "Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine." New England Journal of Medicine 383.27 (2020): 2603-2615.
Sadoff, Jerald, et al. "Safety and efficacy of single-dose Ad26. COV2. S vaccine against Covid-19." New England Journal of Medicine (2021).
Voysey, Merryn, et al. "Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK." The Lancet 397.10269 (2021): 99-111.
熱門點閱》
● 以上言論不代表本網立場,歡迎投書《雲論》讓優質好文被更多人看見,請寄editor88@ettoday.net或點此投稿,本網保有文字刪修權。






我們想讓你知道…在緊急授權造成的科學及倫理兩難情境下,運用所提出之第三期生物相等性臨床試驗不僅可作為二期橋接三期變通做法在科學上的解套,且不需要安慰劑組符合台灣民意風情。